Kepler Community Spotlight: Accelerating Evolutionary Genomics with Subrata Mishra
Tracing evolutionary paths of resistance in different bacterial lifestyles from large population genomic data- When is de novo evolution important?
A Case Study in AI-Accelerated Genomics:
Filtering high-quality, meaningful mutations from large population genomic datasets is a painstaking process — often taking weeks of manual curation and generating numerous intermediate files. From fine-tuning filtering parameters to pinpointing the specific mutations linked to observed phenotypes, each step demands time and precision.
With Kepler AI, this process has been transformed. What once took weeks now takes just minutes. Here’s a case study showcasing how Kepler accelerates and simplifies the analysis of complex genomic data — turning data overload into clear biological insight.
The Challenge:
Microbial populations display two common lifestyles: free-living (planktonic) cells and surface-attached/aggregated communities (biofilms). Biofilm formation is a stress response and planktonic to biofilm transition occurs due to stressors like environmental nutrient limitation and fluctuating energy availability. While we know both types of lifestyles can allow bacteria populations to acquire different evolutionary trajectories, it is unclear if the upregulation of stress response genes in biofilm populations imposes an “evolutionary reserve” i.e., a reduced opportunity for further adaptive mutations compared with planktonic populations exposed to the same fluctuating starvation stress. To test this scenario, we exposed bacterial cells of three types – planktonic cells, a mix of both planktonic and biofilm cells, and only biofilm forming cells to slow degrees of fluctuating starvation. The evolved cells states were then sequenced generating large population genomics data. I have used two studies for reference for this case study (Study 1) and (Study 2) to see the types of comparative data analysis that can be achieved.
Working with large population genomics datasets poses several significant challenges. One of the foremost difficulties lies in selecting appropriate tools for variant calling — different software pipelines (e.g., GATK, FreeBayes, bcftools) vary in accuracy, computational demands, and compatibility, making the choice both technical and strategic. The workflow also generates a vast number of intermediate files, often consuming considerable storage space and complicating file management. Running multiple FASTQ files simultaneously, whether on local systems or high-performance computing (HPC) clusters, can be restricted by memory limits, job scheduling constraints, or software dependencies, further slowing progress. Additionally, visualizing and analyzing such massive datasets in R or other statistical tools is computationally intensive and time-consuming, often requiring multiple optimization steps. Finally, deriving biologically meaningful inferences from the data adds another layer of complexity, as distinguishing genuine evolutionary signals from noise demands rigorous statistical validation and careful interpretation.
Kepler’s 5-Phase Solution:
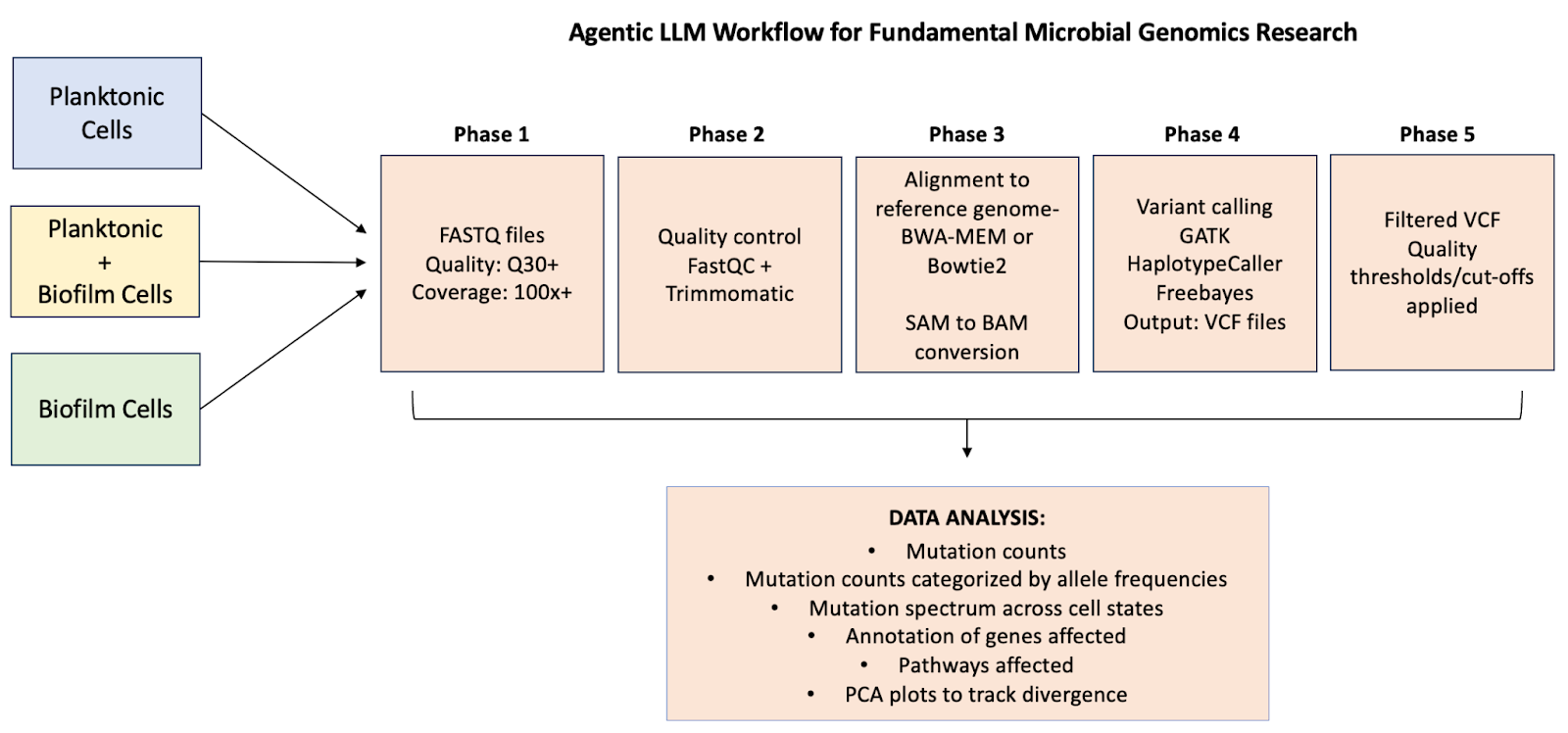
Phase 1: Quality control and generation of BAM files for the three types of bacterial communities
The workflow began by just downloading input FASTQ files- Kepler recommends tools for population genomic datasets and carries out the entire pipeline for obtaining a list of variants. This eliminates the storage of huge intermediate files and weeks of planning and execution.
Input: Simple FASTQ files
Steps: Quality control, alignment, variant calling by multiple tools
Key output: BAM files for all three bacterial community types
Phase 2: VCF files for the three types of bacterial communities
Input: Simple BAM files
Steps: Variant calling by multiple tools for large genomic datasets
Key output: VCF files for all three bacterial community types


Phase 3: Narrowing down to high quality, relevant mutations
Usually, the choice of filtering parameters, the generation of subsequent files can be perplexing and time taking. Kepler sorted and filtered 1000’s of mutations in the VCF files using multiple parameters in a matter of minutes.
Input: VCF files
Steps: Quality controls, depth parameters.
Key output: Filtered VCF files for all three bacterial community types
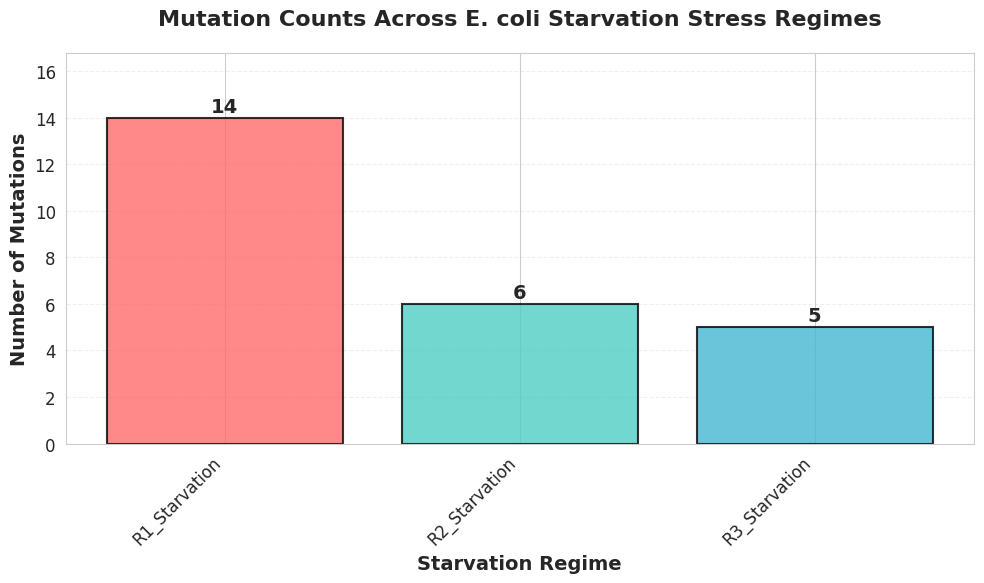
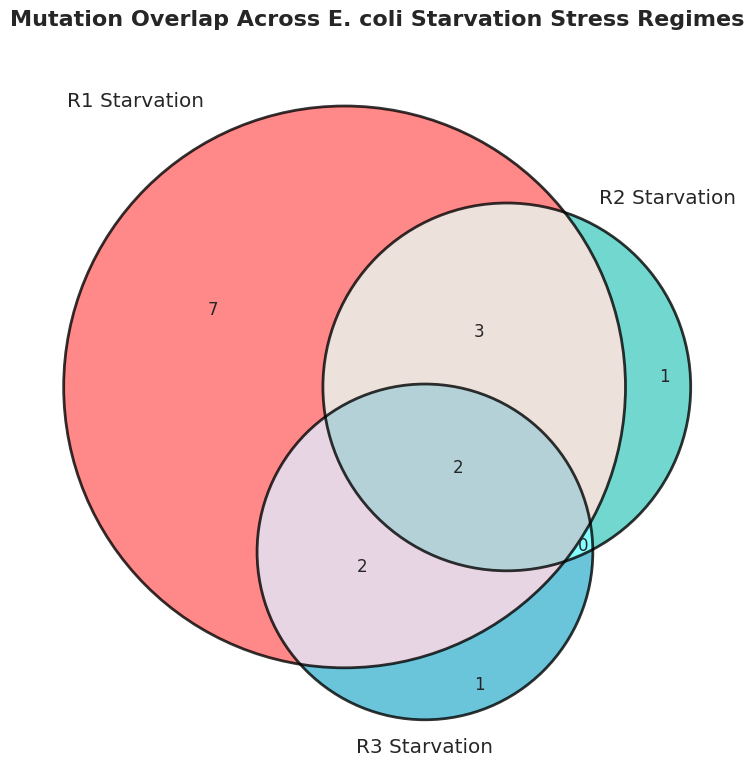

Phase 4: Data analysis of the filtered VCF files
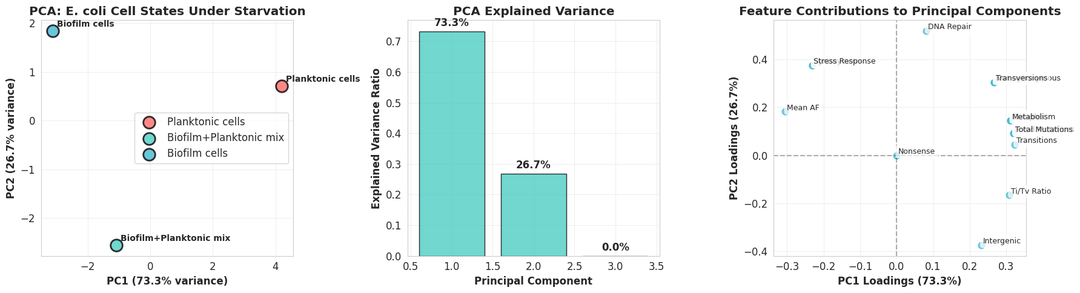
Kepler uses the filtered vcf files for extensive data analysis. From the number of mutations, allele frequencies, overlap among mutations, nature of mutations, and pathways affected to clustering of the three populations, Kepler provides an in-depth data analysis.
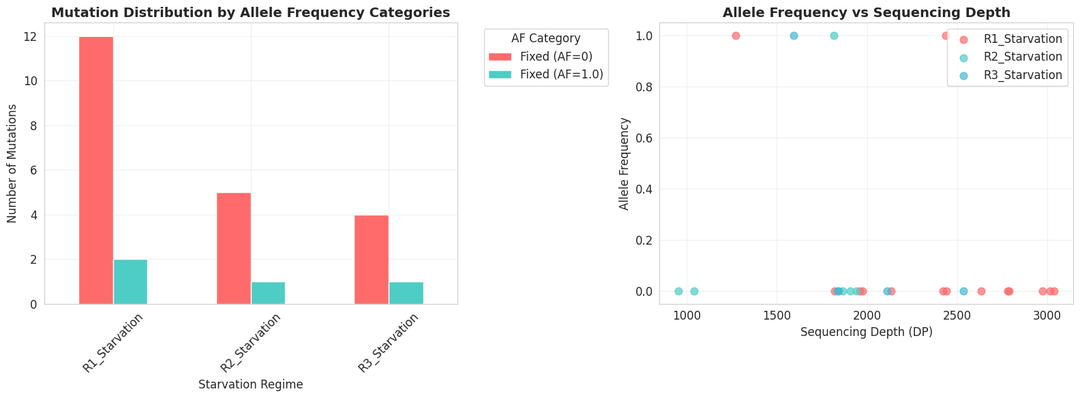
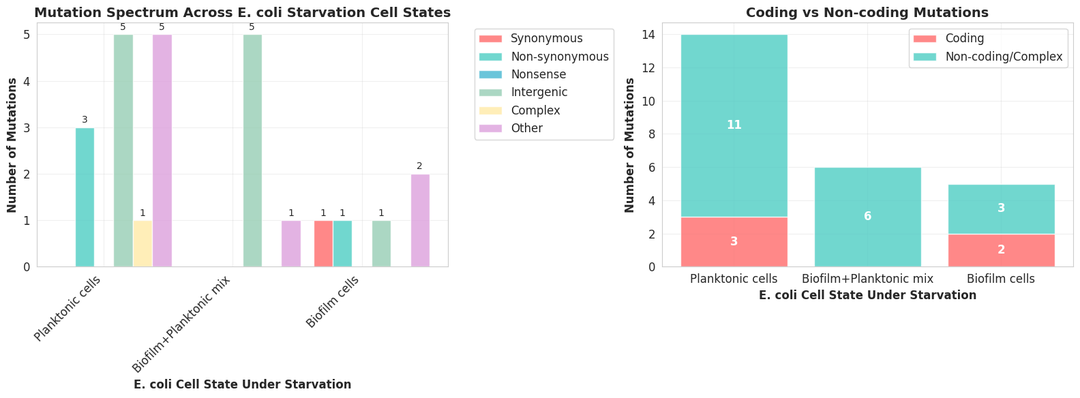
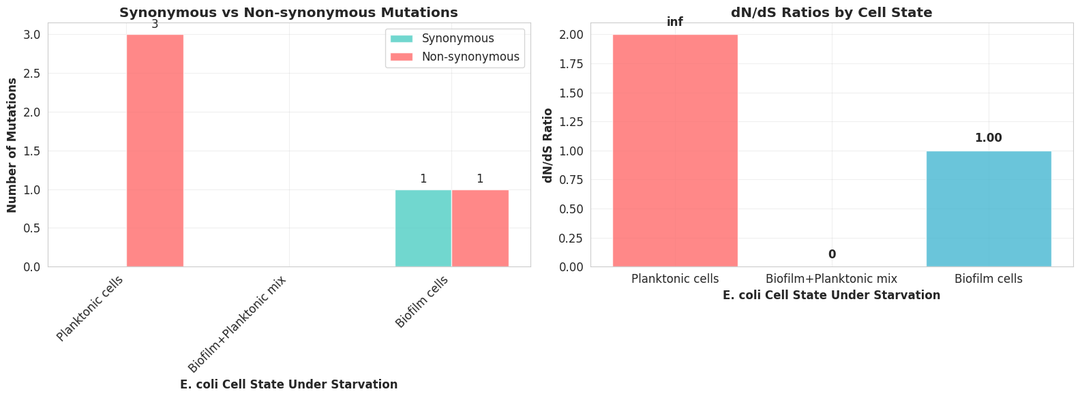
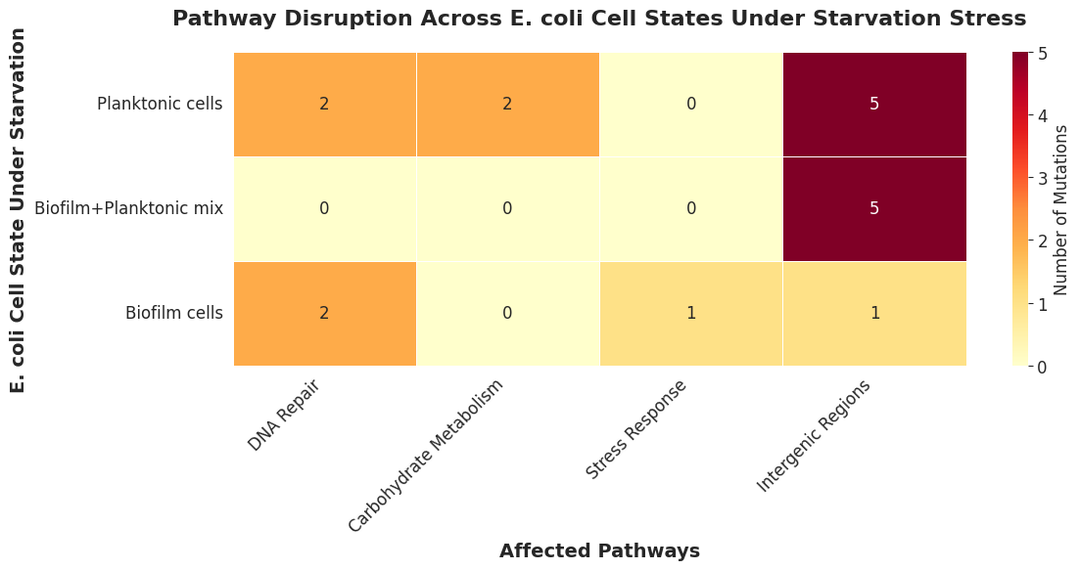
Phase 5: Significance and interpretation of mutation signatures
Finally, the AI integrates all the data to make important inferences and conclusions to the study.
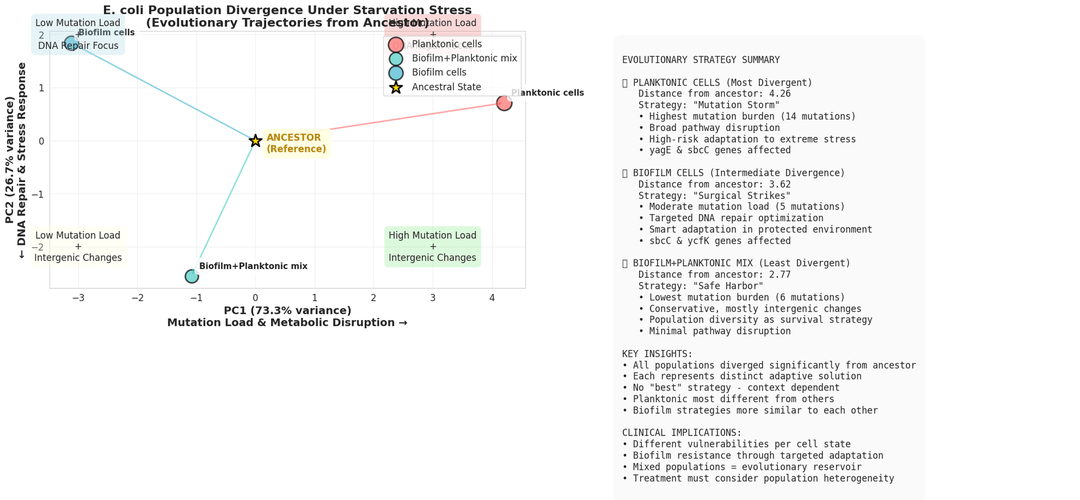
Key Achievements
This case study demonstrates several breakthrough achievements:
- Speed: Complete workflow in minutes versus traditional weeks-long timelines
- Insightful for fundamental science in understanding bacterial resistance and evolutionary trajectories
- End-to-End Automation: From raw data directly to final data files
- Full Transparency: Every step auditable through Kepler's replay functionality

Connect the dots with Kepler
Try out Kepler today, or book a call with us about your organization's use case
Connect the dots with Kepler
Try out Kepler today, or book a call with us about your organization's use case
